
Infračervená spektroskopie
Infračervená spektroskopie , krátká IR spektroskopie a až do šedesátých let minulého století, nazývaná infračervená spektroskopie , je fyzikální analytická metoda , která s infračerveným zářením (vlnová délka: 800 nm až 1 mm) funguje. Metoda je jednou z metod molekulární spektroskopie, které jsou založeny na excitaci energetických stavů v molekulách.

IR spektroskopie se používá pro kvantitativní stanovení známých látek, které jsou identifikovány pomocí referenčního spektra, nebo pro strukturální objasnění neznámých látek.
Podobné molekulární spektroskopické metody jsou Ramanova spektroskopie , která také poskytuje informace o vibracích v infračerveném rozsahu, a UV / VIS spektroskopie ve vyšším frekvenčním rozsahu.
V dnešní době se používají především Fourierovy transformační infračervené spektrometry (FTIR spektrometry). Ve srovnání s disperzními spektrometry, které byly použity v minulosti, tyto nabízejí řadu rozhodujících výhod. Například spektrometry FTIR mají výrazně vyšší poměr signálu k šumu a vyžadují výrazně kratší doby měření. Výsledky infračervené spektroskopie jsou uvedeny v infračervené spektrografii.
varianty
Ze spektroskopického hlediska se rozlišuje blízký infračervený (NIR; vlnové číslo : 12500–4000 cm −1 ; vlnová délka: 0,8–2,5 µm, viz blízká infračervená spektroskopie ), střední nebo klasický (normální) infračervený ( MIR; vlnové číslo: 4000–400 cm −1 vlnová délka: 2,5–25 µm) a vzdálené infračervené záření (FIR; vlnové číslo: 400–10 cm −1 ; vlnová délka: 25–1 000 µm), protože díky tomu lze pozorovat různé jevy k absorpci v příslušné oblasti jsou. Absorpce v rozmezí FIR vede hlavně k rotaci celých molekul. V rozsahu MIR a rozsahu NIR dochází k excitaci oscilace atomů nebo skupin atomů na jejich molekulárních vazbách, přičemž v oblasti NIR jsou detekovatelné takzvané harmonické řady MIR (zejména vazeb CH, OH a NH).
Mid -infračervená spektroskopie - často označovaná jednoduše jako IR spektroskopie - je účinná technika v chemické analýze organických látek. Umožňuje přímá prohlášení o přítomnosti a případně koncentraci infračerveně aktivních funkčních skupin . Blízké infračervené spektroskopie (NIRS) je (často, zejména v aplikacích pro rychlé hodnocení látek a směsí procesní analýzy ), používané, ale k počítačovým chemometrické modely spoléhají s průvodním, náhodně nesprávné referenční analýzy.
V IR spektrech, na rozdíl od UV spektra, často není míra propustnosti excitačního záření absorpce, ale spíše přenos . Přenos je zakreslen na svislé ose a zvyšuje se směrem nahoru - oblasti s nízkým přenosem infračerveného záření vedou k vychýlení směrem dolů. Používá se však také reprezentace jako absorpční spektrum, zejména u technologie ATR.
Princip měření
Absorpce IR záření
Když je látka ozářena elektromagnetickými vlnami , jsou absorbovány určité frekvenční rozsahy. Pokud jde o energii, infračervené záření leží v rozsahu úrovní rotace malých molekul a úrovní vibrací molekulárních vazeb, tj. To znamená, že absorpce způsobí, že vazby vibrují . Jsou viditelné ve formě průhybů v měřeném spektru (diagram). Protože energie nebo frekvence k tomu potřebné jsou charakteristické pro příslušné vazby, lze materiály identifikovat také tímto způsobem. IR spektroskopie proto objasňuje strukturu.
Interakce mezi elektromagnetickým zářením a molekulou může nastat pouze tehdy, je -li v molekule pohybující se elektrický náboj. To je vždy případ, kdy má molekula buď měnitelný nebo indukovatelný dipólový moment (IR aktivní). V molekulách s vibracemi symetrickými ke středu symetrie nedochází ke změnám v dipólovém momentu (IR neaktivní). Takové „zakázané“ vibrace jsou často Ramanovy aktivní .
Nejjednodušším případem je diatomická molekula. V případě polyatomických molekul se základní vibrace překrývají. V souladu s tím je vidět řada absorpčních pásem, které je třeba interpretovat.
Mechanický model


Nejjednodušší představitelný model, který lze použít k vysvětlení vibračních a rotačních buzení, je klasický model interakce permanentního elektrického dipólu v elektromagnetickém poli. Později mohou lepší modely, jako je kvantově mechanický model, také popisovat molekuly bez trvalého dipólového momentu.
Mezi atomy a jejich sousedy existují atraktivní a odpudivé interakce. Proto je optimální vzdálenost vazby v molekule na minimu potenciální funkce. Mechanicky si to lze představit, jako by atomy byly navzájem spojeny pružinami. Síla, která je potřebná k vychýlení pružiny, je popsána Hookeovým zákonem pružin . Pokud někdo přivede takovou molekulu do elektrického pole, jaké existuje například v deskovém kondenzátoru, molekula se nejprve zarovná se svým dipólovým momentem podél elektrického pole a za druhé zvýší svoji vzdálenost vazby. Pokud je nyní aplikováno střídavé napětí nebo je molekula buzena elektromagnetickou vlnou, funkční skupiny „visící“ na vazbách začnou vibrovat a otáčet se. Mechanistický model má pouze omezené použití, protože však například nevysvětluje, proč jsou pro excitaci infračerveného záření povoleny pouze diskrétní energie a proč molekuly bez trvalého dipólového momentu také vykazují absorpci infračerveného záření.
Kvantově mechanický model

Stejně jako v modelu klasické mechaniky je základem kvantově mechanického modelu buzení vibrací a rotace potenciální funkce. Minimum potenciální funkce lze aproximovat parabolou. Taková parabola vyplývá z integrace Hookeova zákona pružin.
Pokud je molekula nyní buzena elektromagnetickým zářením, mohou být valenční vazby uvnitř molekuly nastaveny na oscilace a rotace, pokud je energie dostatečná na to, aby ji zvedla ze základního stavu do prvního stavu excitované oscilace. K určení této energie musí být pro tento potenciál vyřešena Schrödingerova rovnice . Po oddělení relativního pohybu atomových jader a elektronů ( Born-Oppenheimerova aproximace ) výsledkem řešení Schrödingerovy rovnice je vztah mezi požadovanou energií, pevností vazby ( k ) a sníženou hmotností ( μ ). Na rozdíl od klasického harmonického oscilátoru je v kvantově mechanickém případě vibrační energie kvantována kvantovým číslem vibrací v .
Schrödingerova rovnice:

Řešení Schrödingerovy rovnice:

Průběhy
V anorganických a organických látkách dochází k mechanickým vibracím, když je absorbováno záření z infračerveného rozsahu. Podle R. Mecke lze molekulární vibrace rozdělit na dva typy:
- Protahovací vibrace (roztahovací vibrace): vibrace podél osy vazby dvou atomů nebo částí molekuly v důsledku prodloužení nebo stlačení vazby
-
Deformační vibrace : vibrace při deformaci úhlu vazby
- v rovině (ohybové / ohybové vibrace):
- mimo rovinu (torzní / naklápěcí vibrace) - většinou kolmo na rovinu vazby
Symetrický úsek
(angl. Symetrical stretching )
Antisymetrický streč
(angl. Antisymetrické strečink )
Vibrace při ohybu
( nůžky nebo ohýbání v angličtině )
Houpací vibrace
(angl. Rocking )
Houpací oscilace
(angl. Wagging )
Torzní vibrace
( kroucení nebo kroucení v angličtině )






Protože jsou zvláště charakteristické molekulární vibrace určitých atomových skupin v rozmezí 4000–1500 cm −1 , je IR spektroskopie vhodná pro stanovení určitých strukturních prvků, zejména funkčních skupin zkoumané molekuly.
Spektrum rotačních vibrací
Ideální spektrum rotace-oscilace

Chlorovodík (HCl) je diatomická molekula s výrazným dipólovým momentem. Zjednodušeně lze na molekulu HCl pohlížet jako na lineární vrchol . Stejně jako v čisté rotační spektroskopii lze k popisu spekter použít model tuhého rotátoru . Pro lineární molekulu platí speciální pravidlo výběru pro rotační přechod:
- ,

kde představuje rotační kvantové číslo. Pokud je dipólový moment rovnoběžný s hlavní osou otáčení (jako je tomu například u čpavku ), bylo by to také možné. Zvláštní pravidlo výběru pro oscilační přechod harmonického oscilátoru je:



Přechody mezi různými úrovněmi otáčení ( ) základního stavu vibrací ( ) a úrovněmi otáčení ( ) excitovaného stavu vibrací ( ) lze použít ve dvou skupinách, R-větev (vpravo) pro přechody a P-větev (vlevo ) u přechodů jsou a jsou na obrázku načrtnuty dělené. Protože HCl není povoleno, nezobrazí se žádná Q větev. Schéma energetické úrovně je zobrazeno na obrázku výše. Délka šipky jasně ukazuje, že každý přechod je zásobou energie






vyžaduje, kde je konstanta otáčení s jednotkou . To zahrnuje moment setrvačnosti molekuly vzhledem k ose rotace.



Následující platí pro energetický rozdíl k sousednímu přechodu

Z tohoto důvodu, v ideálním otáčení vibračního spektra (dolní panel) vrcholy mají konstantní vzdálenost 2 B . V experimentu jsou vrcholy rozmístěny mírně odlišně, protože model:
- anharmonicita vibrací
- rozšířená atomová vzdálenost odstředivými silami (přiblížení: tuhý rotátor )
- a je pozorována interakce vibračního a rotačního pohybu.
Skutečné spektrum oscilace rotace
Skutečné spektrum oscilace otáčení se odchyluje od ideálu kvůli omezením použitých modelů. Vzdálenosti mezi píky (výchylka amplitudy v diagramu, nazývaná také pásma ) nejsou přesně 2 B , jako v ideálním případě , ale s přibývajícím vlnovým počtem ve větvi R se zmenšují, ale ve větvi P se zvětšují interakce rotace a oscilace. Opačný diagram ukazuje zaznamenané spektrum chlorovodíku v plynné fázi. V jemné struktuře spektra je jasně vidět zdvojnásobení všech pásů v důsledku dvou různě inertních izotopů chloru.
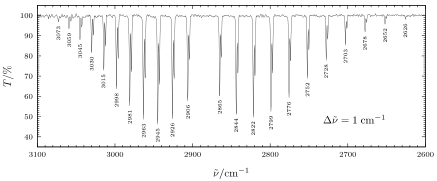
Spektrum rotačních vibrací plynného chlorovodíku při pokojové teplotě.

Spektrum rotačních vibrací plynného oxidu uhličitého měřeno při pokojové teplotě s různým rozlišením.


Poloha a vyhodnocení absorpčních pásem IR
Silnější chemické vazby a atomy menší hmotnosti způsobují absorpční maxima v IR spektru při velkých vlnových číslech (vysoká energie), zatímco velké hmotnosti způsobují absorpční maxima IR při malých vlnových číslech (nízká energie) (viz deuterace ). Energie je úměrná druhé mocnině trvalého dipólového momentu. Polární molekuly proto poskytují intenzivní rotační přechody. Nicméně, jestliže jeden porovnává amplitudy píků jedné molekule se navzájem, je patrné, že síla přechodů zpočátku rychle narůstá s rostoucí J , prochází maximem a nakonec snižuje opět pro velké J. Důvodem je to, že síla odráží degenerace různých rotačních stavů a čísla obsazení rotačních úrovní v počátečním stavu. Stupeň degenerace se zvyšuje s rostoucím J , což vede k vyšší energii. Na druhou stranu počet povolání klesá s rostoucí energií, což nakonec vede ke snížení intenzity záření.

IR spektra jsou interpretována tak, aby bylo možné odvodit molekulární tvar z křivky měřeného IR spektra . Měří se různé vibrační varianty molekul a lze vyvodit závěry o přítomnosti určitých strukturních prvků. Typické IR spektrum se pohybuje od 4000 cm −1 do 400 cm −1 ( vlnové číslo ). Číslo vlny je udáváno v poněkud neobvyklé jednotce cm −1 . To však má tu výhodu, že číslo vlny a energie buzení jsou proporcionální. Energii dává vlnové číslo vynásobené Planckovým kvantem a rychlostí světla . V IR spektru každá molekula zanechává typický vzor absorpčních pásem. Délka těchto pásem se obvykle nehodnotí číselně, ale hodnotí se pouze s atributy jako „ silný “ nebo „ nepříliš intenzivní “. Od asi 1 500 cm −1 směrem dolů se přiřazování jednotlivých pásem k určitým molekulárním vibracím stává výrazně obtížnějším, protože počet možných překrytí v důsledku vibrací rámce je velmi vysoký. Tato takzvaná oblast otisků prstů by v ideálním případě měla poskytovat charakteristické spektrum pro každý typ molekuly. Ve skutečnosti lze předpokládat, že některé rozdíly nelze vyjádřit přesností měření spektrální analýzy. V této oblasti však existují i některé pásy, které jsou charakteristické pro určité strukturální prvky. Páry enantiomerů vykazují zcela identická spektra, pokud není použito lineárně polarizované infračervené světlo. Pokud neexistují žádné čisté látky, ale směsi , IR spektrum obsahuje informace o strukturních prvcích všech složek směsi. S odpovídajícím spektrem s vysokým rozlišením však mohou být jednotlivé složky stále detekovány na základě charakteristických absorpcí určitých skupin látek nebo kombinace několika absorpcí. Často se provádí přímé srovnání se spektrem čisté látky.


Individuální přiřazení pásem je založeno na tabulkových empirických datech. Přesnou polohu pásma je možné posunout na vyšší nebo nižší vlnovka díky chemickému prostředí příslušné atomové skupiny, což umožňuje vyvodit další závěry o blízkosti určitých strukturních prvků. Při interpretaci poloh pásem je však třeba vzít v úvahu, že různé optické efekty mohou také vést k posunům pásma, např. B. interferenční a vyrovnávací efekty a také polarizace hmoty světlem. To ztěžuje interpretaci takových posunů.
| Označení podle Mecke / atomová skupina | Rozsah čísel vln v cm −1 |
komentář |
|---|---|---|
| ν (–C - H x ) | 2850-3200 | ν = roztahovací vibrace; × = 1… 3, počet vázaných atomů vodíku |
| δ (–C - H x ) | 1400 | δ = deformační vibrace; × = 1… 3, počet vázaných atomů vodíku |
| -C = C | 1650 | |
| –C≡C | 2200-2500 | |
| ν (-OH) | 3200-3600 | V souvislosti s vodíkovými vazbami se pásy často nacházejí ve velkém spektrálním rozsahu, například v případě kapalné vody |
| ν (-OH) | 2500-3000 | v karboxylových skupinách |
| -C = O | 1700 | |
| ν (–C≡N) | 2200-2260 | Protahovací vibrace, např. B. v ABS nebo SAN (viz obrázek) |
| ν (–NH) | 3100-3500 | |
| -NE 2 | 1500 | |
| -C-X | <1500 | kde X je atom halogenu , je |
| ν jako (O = C = O) | 2349 | Oxid uhličitý. |
Techniky IR spektroskopie
přenos
Jednou z nejčastěji používaných metod je měření propustnosti vzorku. Za tímto účelem se vzorek ozařuje infračerveným zářením a měří se podíl měřicího záření, které do vzorku proniklo. Pokud má vzorek IR-aktivní vazby nebo skupiny molekul, jsou v přenosovém spektru vidět oblasti nižší intenzity (absorpční pásy). Podle Lambert-Beerova zákona (mimo jiné) se intenzita pásma zvyšuje s tloušťkou vzorku (je však třeba poznamenat, že tento zákon obecně platí pouze přibližně, protože není kompatibilní s Maxwellovými rovnicemi Technologie měření více resp. méně velké odchylky. Podrobná vysvětlení a možná řešení najdete v). Kromě toho se část záření také odráží ; odpovídající části mají obvykle účinek zvýšeného sníženého přenosu v celém spektru a lze je do značné míry odstranit korekcí základní linie.
V závislosti na tvaru vzorku (plynný, kapalný, pevný) existují speciální měřicí techniky, které se liší typem přípravy vzorku a měřicími buňkami (držák vzorku s více či méně komplikovanými drahami paprsku). Nejjednodušší uspořádání měření odpovídá jednoduchému držáku vzorku, který fixuje těleso (např. Fólii nebo desku) v dráze paprsku spektrometru. U vzorků s paralelními rozhraními (jako film) však interference vznikají z více odrazů ve vzorku (viz také Newtonovy prstence ), které v závislosti na tloušťce a rovnoběžnosti rovnají spektrum, ale interference lze také použít k určení tloušťka vrstvy .


Široce používanou technikou pro měření pevných vzorků je vložení pevného tělesa do IR transparentního nosného materiálu (také známého jako matrice). Hlavně anorganické soli , jako jsou halogenidy v alkalických kovů se používají , společná matice je bromid draselný (KBr). K měření pevných látek se suchý práškový bromid draselný (KBr je hygroskopický ) smíchá s některou vzorkovou látkou a intenzivně se rozemele. Tato směs se potom lisuje do tablety v ocelovém válci za vakua a vysokého mechanického tlaku. Při tomto tlaku se bromid draselný stává plastickým a studený tok (obvykle také opticky) vytváří průhledné „pelety“. Ty pak lze jednoduše umístit do dráhy paprsku.
Kapalné vzorky lze umístit mezi dvě infračervené transparentní desky vyrobené z monokrystalů halogenidů alkalických kovů nebo zřídka průhledných polymerových desek. To lze provést staticky nebo dynamicky ve formě průtokové jednotky. Plynné vzorky jsou obvykle umístěny do speciálních kyvet .
Měření tloušťky vrstvy

Polypropylenová fólie s interferencemi (tloušťka přibližně 38 µm)
|

Protože dochází k interferenci s rovinně rovnoběžnými tenkými vzorky, je skutečné spektrum superponováno sinusovým vlnovým sledem. Tloušťku fólie lze vypočítat z polohy maxim.

se na indexu lomu vzorku a počet maxim ve spektrální oblasti mezi vlnových délkách a .




Rušení ve fóliích můžete odstranit jejich pomačkáním nebo zdrsněním. Přitom se však zvýší podíl difúzního odrazu. Alternativní metodou je takzvaná disperzní analýza. Naměřené spektrum je modelováno na základě vlnové optiky a teorie disperze a jsou získány optické konstanty, tloušťka vrstvy a parametry oscilátoru. Tímto způsobem lze obecně odstranit celou řadu optických efektů.
Christiansenův efekt
Christiansen účinek (dle Christian Christiansen ) je artefakt IR přenos měření látek vázaných v bromidu draselného nebo jiné matrici (tzv pelet). Projevuje se především prostřednictvím asymetrických pásem ve spektru. Vysokofrekvenční hrana ukazuje velmi strmý průběh (často vyšší než základ spektra). Nízkofrekvenční bok má naopak tendenci vykazovat plochý, protáhlý kurz. Příčina spočívá v anomální disperzi v blízkosti silných absorpčních pásem . Účinek je však pozorován pouze u pelet, ve kterých je zkoumaná látka příliš hrubozrnná, nebo u vzorků s větším rozdílem v indexu lomu mezi zkoumanou látkou a materiálem matrice. To znamená, že účinek lze minimalizovat nebo mu lze zabránit, pokud se velikost látky zmenší mletím.
Odraz a absorpce



Další metodou je (vnější) odraz infračerveného záření na hladkém povrchu. Metoda využívá skutečnosti, že míra odrazu vzorku závisí na komplexním indexu lomu vzorku (viz Fresnelovy vzorce ). V oblasti center oscilace se absorpce zvyšuje, což je v komplexním indexu lomu zastoupeno zvýšením extinkčního koeficientu ( pomyslná část indexu lomu). Toto zvýšení je doprovázeno změnou indexu lomu kolem absorpčního pásma, která je v první aproximaci úměrná tomuto zvýšení. Zvýšení indexu lomu i indexu absorpce vede ke zvýšenému odrazu vzorku pro toto frekvenční pásmo a k pásmu v odrazovém spektru. U slabých oscilátorů, jako jsou ty, které se nacházejí převážně v organických materiálech, převládá vliv změny indexu lomu, což je důvod, proč spektra ukazují tvary pásů podobné derivátům , zatímco vliv absorpčního indexu se zvyšuje se zvyšující se silou oscilátoru a určuje pásmo tvar. Důvod pro tvary pásů podobných derivátům spočívá v anomální disperzi v blízkosti absorpčních center materiálu, tj. Index lomu se zvyšuje s vlnovou délkou místo klesání jako obvykle. To vede ke změně odrazivosti a tím k pozorovaným zkresleným pásmům. Rostoucí vliv absorpčního indexu naopak vede k tvarům pásů, které dočasně připomínají absorpční pásy, než se z nich stanou velmi široká pásma s plošinami v blízkosti 100% odrazu, jejichž snížení pak indikuje přítomnost slabších oscilátorů .
Tato technika je vhodná mimo jiné pro měření silně absorbujících vzorků. Hlavní nevýhodou metody je, že se odrazová spektra výrazně liší od spektra přenosu. Nedávno byla pro reflexní spektra organických materiálů navržena nová kvantitativní metoda hodnocení, což je podobně jednoduché hodnocení jako u absorpčních spekter na základě Lambert-Beerova zákona . Tato metoda se proto nazývá infračervená refrakční spektroskopie. Pro reflexní spektra anorganických materiálů se silnými oscilátory se pro výpočet absorpčního spektra vzorku používá buď disperzní analýza nebo Kramersova-Kronigova transformace (KKT).
Kromě výše popsané metody je vhodná další metoda odrazu, zejména pro existující tenké vrstvy, a adsorbáty, takzvaná technika " infračervené odrazové absorpční spektroskopie " (angl.: Infračervená odrazová absorpční spektroskopie , IRRAS, také IRAS). Zde se infračervené záření odráží na kovovém povrchu (vnějším), na kterém se nachází zkoumaná vrstva. Vzhledem k velmi vysoké odrazivosti kovové vrstvy (> 90%) je infračervené záření prakticky dvakrát vedeno vzorkem. Navíc, podobně jako povrchová plazmonová rezonanční spektroskopie , je pole na kovovém povrchu posíleno, což činí metodu citlivou i pro monovrstvy , tj. Tenké vrstvy s tloušťkou vrstvy několika angströmů ; detekce monovrstev je také možná s jinými měřicími technikami infračervené spektroskopie. Měření se obvykle provádí při kvazi-pastevním dopadu, tj. Pod úhlem dopadu přibližně 80 ° od kolmice. Kromě toho lze citlivost metody zvýšit použitím p-polarizovaného záření . I v tomto případě lze spektra kvantitativně vyhodnotit pomocí metody disperzní analýzy.
Difúzní odraz

Pomocí difúzní reflexní spektroskopie (DRIFTS) lze zkoumat hrubé pevné látky a prášky, aniž by byly tyto lisovány do matrice (viz měření transmise). Infračervený paprsek je nasměrován na vzorek a vzhledem k drsnému povrchu již není směrován (jako zrcadlo), ale difúzně se odráží . Odražené záření je opět svázáno speciálním zrcadlovým uspořádáním, polokulovitým nad vzorkem (viz konkávní zrcadlo ) a vedeno k detektoru.
Vzhledem k difuznímu odrazu jsou složky záření nerovnoměrně rozloženy a výsledné spektrum má zvláštnosti, které pouze ve velmi omezené míře umožňují přímé srovnání se spektry jiných IR technologií. Pro hodnocení (kvalitativní a kvantitativní) lze spektrum převést na absorpční spektrum pomocí funkce Kubelka-Munk .
Oslabený celkový vnitřní odraz (ATR)

V infračervené spektroskopii ATR je záření vedeno v úplném odrazu v odrazovém prvku ( takříkajíc světelném vlnovodu ). Na rozhraní elementu se tvoří evanescentní pole, jehož amplituda se exponenciálně rozpadá kolmo na rozhraní ( evanescentní vlna ). Pokud je za rozhraním vzorek, evanescentní pole interaguje se vzorkem, tj. Určité frekvenční rozsahy mohou být absorbovány vzorkem. Absorbované oblasti nyní chybí ve spektru odraženého paprsku, který je veden k detektoru. Vzhledem k tomu, že se pole velmi rychle rozpadá, je třeba vzorky pro měření přivést velmi blízko k povrchu, aby se dosáhlo intenzit pásma, které lze ještě vyhodnotit. Měřená oblast obvykle pokrývá pouze první mikrometry vzorku. Výsledkem jsou podobná spektra jako v případě transmisní spektroskopie, které však mají ve srovnání rozdíly v intenzitě závislé na vlnové délce. Tato metoda je vhodná pro tuhé a kapalné vzorky se srovnatelně slabými absorpcemi, které jsou doprovázeny pouze malými změnami indexu lomu, jinak již elektrické pole ve vzorku neplyne.
emise

V infračervené emisní spektroskopii (angl. Infračervená emisní spektroskopie , IRES) vzorku je studováno emitované infračervené záření. To znamená, že na rozdíl od výše popsaných metod absorpční spektroskopie není vzorek ozařován vysokoteplotním světelným zdrojem a srovnáván s referenčním spektrem bez vzorku, ale samotný vzorek slouží jako zdroj záření. Získaná emisní spektra ukazují podstatně méně podrobností s ohledem na rotační spektra a intenzita a tvar pásma jsou více závislé na teplotě.
Měření vzorků bez referenčního spektra je možné, protože molekula, která je ve stavu excitované oscilace, může s určitou pravděpodobností vyzařovat infračervené záření. Naměřená emisní pásma obvykle odpovídají přechodu z excitovaného do základního stavu. K vyzařování může docházet jak přítomností (prostřednictvím indukované emise ), tak v nepřítomnosti (prostřednictvím spontánní emise ) dopadajícího elektromagnetického záření. Při pokojové teplotě je však počet molekul, které jsou v excitovaném stavu, relativně malý (méně než 1% počtu v základním stavu, vztaženo na mezní hodnotu při 1 000 cm −1 ), a proto je výrazně menší než v případě absorpce. Aby byl získán silnější signál, vzorek se obvykle zahřívá (obvykle nad 100 ° C) a měří se vyzařované záření (viz černé těleso ). Navíc na základě součinu Boltzmannova faktoru a pravděpodobnosti přechodu lze očekávat pouze velmi nízké energie záření, takže IRE spektroskopie byla po dlouhou dobu velmi složitá a vykazovala pouze nízkou citlivost. S vylepšenými interferometry a novými citlivými detektory lze nyní rutinně zaznamenávat emisní spektra.
IRE spektroskopie se používá mimo jiné pro zkoumání barev , roztavených solí a křemičitanů a také katalytických reakcí na tenkých vrstvách a adsorbovaných druhů na pevných látkách. Tato metoda nabízí výhody především pro měření in situ při vysokých teplotách, protože zde, na rozdíl například od reflexních technik, je překrývání absorpčních pásem s emisními pásy zanedbatelné. IRE spektroskopii lze také použít k měření velkých objemů plynu, například při procesní analýze .
Měření více vrstev nebo obecně větších objemů může být problematické, protože zde probíhají absorpční a emisně reabsorpční procesy a ovlivňují naměřená spektra. Mezi další parametry ovlivňující spektrum patří geometrie vzorku, volba radiačního pozadí, rozptýlené záření a, jak již bylo řečeno, teplota samotného vzorku.Z těchto důvodů dokonce i IR emisní spektra s vysokým signálem-šumem poměry jsou velmi složité a obtížně se hodnotí. V literatuře byly proto uvedeny různé metody ke kompenzaci interference.
Viz také
- Povrchová chemie
- Alternativní zákaz
- Globar
- Princip Franck-Condon
- Journal of Near Infrared Spectroscopy
literatura
- Helmut Günzler, Hans-Ulrich Gremlich: IR spektroskopie: úvod . 4. vydání. Wiley-VCH, Weinheim 2003, ISBN 978-3-527-30801-9 .
- M. Hesse, H. Meier, B. Zeeh: Spektroskopické metody v organické chemii . 6. vydání. Thieme, Stuttgart 2002, ISBN 3-13-576107-X .
- Bernhard Schrader : Infračervená a Ramanova spektroskopie . VCH, 1995, ISBN 3-527-26446-9 .
webové odkazy
- Základy a technologie infračervené spektroskopie
- Spectral Database for Organic Compounds, SDBS (anglicky)
- Sbírka různých IR spekter
- IR spektroskopie a organická chemie (anglicky)
- Spektra rotačních vibrací v Chemgapedia
- Adresář databází a referencí ETH v Curychu pracuje s IR spektry
- Adresář databází a referenčních prací ETH v Curychu se vzdálenými IR spektry
- Adresář databází a referenčních prací ETH v Curychu se spektry blízkého IR
Individuální důkazy
- ↑ BGR infračervená spektroskopie. Citováno 17. července 2008 .
- ↑ a b Alexander Erlich: Infračervená spektroskopie s Fourierovou transformací. ( Memento z 11. prosince 2014 v internetovém archivu ) (Příklady spekter IRES; PDF; 2,6 MB), 8. července 2010.
- ↑ Helmut Günzler, Hans-Ulrich Gremlich: IR spektroskopie: Úvod . 4. vydání. Wiley-VCH, Weinheim 2003, ISBN 978-3-527-30801-9 , s. 165-240 .
- ^ Thomas Günter Mayerhöfer, Susanne Pahlow, Jürgen Popp: The Bouguer-Beer-Lambert law: Shining light on the temný . In: ChemPhysChem . n / a, n / a, 14. července 2020, ISSN 1439-4235 , doi : 10,1002 / cphc.202000464 .
- ↑ Thomas G. Mayerhöfer, Susanne Pahlow, Uwe Hübner, Jürgen Popp: Odstranění interferenčních efektů z infračervených spekter-interferenční proužky se znovu vrátili . In: Analytik . páska 145 , č. 9 , 4. května 2020, ISSN 1364-5528 , s. 3385-3394 , doi : 10,1039 / D0AN00062K ( rsc.org [přístup 10. ledna 2021]).
- ↑ C. Christiansen: Vyšetřování optických vlastností jemně rozdělených těl - první komunikace . In: Annals of Physics and Chemistry . 23, 1884, s. 298-306.
- ↑ C. Christiansen: Vyšetřování optických vlastností jemně rozdělených těles - druhá komunikace . In: Annals of Physics and Chemistry . 24, 1885, s. 439-446.
- ↑ CV Raman : Teorie experimentu Christiansen . In: Proč. Ind. Acad. Sci. 29, 1949, s. 381-390.
- ↑ a b c d (PDF) Vlnová optika v infračervené spektroskopii. Přístup 21. srpna 2021 .
- ^ Thomas G. Mayerhöfer, Vladimir Ivanovski, Jürgen Popp: Infračervená refrakční spektroskopie . In: Aplikovaná spektroskopie . 18. srpna 2021, ISSN 0003-7028 , s. 000370282110367 , doi : 10,1177 / 00037028211036761 ( sagepub.com [přístup 21. srpna 2021]).
- ^ Thomas Mayerhöfer: Vlnová optika v infračervené spektroskopii . 2021, doi : 10.13140 / RG.2.2.14293.55520 ( rgdoi.net [přístup 26. dubna 2021]).
- ↑ a b W. Suetaka: Surface Infrared and Raman Spectroscopy: Methods and Applications . Springer, New York 1995, ISBN 0-306-44963-3 , s. 163 ff .
- ^ S. Zhang, FS Franke, TM Niemczyk: Emisní spektroskopie . In: Francis Mirabella, Mirabella (Ed.): Modern Techniques in Applied Molecular Spectroscopy . Wiley & Sons, 1998, ISBN 0-471-12359-5 , s. 323–376 (úvod s popisy různých technik).
- ↑ K. Molt: Záznam infračervených emisních spekter tenkých vrstev na kovových površích pomocí počítačově spřaženého mřížkového spektrometru . In: Fresenius 'Journal for Analytical Chemistry . páska 308 , č. 4 , 1981, s. 321-326 , doi : 10,1007 / BF00516246 (s odkazem na G. Herzberga: Spektra diatomických molekul. Van Nostrand, Princeton 1950.).
- ^ Francis M. Mirabella: Moderní techniky v aplikované molekulární spektroskopii . Wiley-IEEE, 1998, ISBN 978-0-471-12359-0 , 9. emisní spektroskopie, str. 323-377 .